Express RNA-Seq Services Overview
Express RNA-Seq is a next-generation sequencing (NGS) based mRNA profiling service designed to deliver rapid, high-quality transcriptome data. This approach selectively captures polyadenylated transcripts and uses a transposase-based tagmentation workflow to enable efficient, accurate gene expression analysis.
During library preparation, mRNA is reverse-transcribed using an oligo-dT primer. The resulting cDNA is simultaneously fragmented and tagged with sequencing adapters in a single enzymatic reaction. This streamlined workflow accelerates project completion while maintaining high data quality and integrity.
With results delivered in as little as 4 weeks, this service is ideal for projects requiring rapid turnaround, including time-sensitive studies, screening experiments, and high-throughput gene expression profiling.
Express RNA-Seq Highlights:
- Ultra-fast turnaround time (4 weeks)
- Developed for mRNA sequencing (polyA-enriched)
- High-throughput format to reduce batch-to-batch variation
- End-to-end service from RNA isolation to data interpretation
- 15–20 million paired-end 50 bp reads (PE50)
- Publication-ready figures included
From RNA isolation and library preparation through sequencing and data analysis, we provide comprehensive end-to-end support to deliver publication-ready results. For deeper biological insights, RNA-Seq can be combined with our industry-leading ChIP-Seq or ATAC-Seq, enabling integrated characterization of gene expression programs and regulatory chromatin architecture within your specific samples or experimental model.
Why Choose Active Motif for Express RNA-Seq?
End-to-End Expertise in Gene Regulation. As a leader in epigenetics research, Active Motif combines transcriptomics with deep knowledge of chromatin biology to deliver biologically meaningful insights.
High-Quality Data You Can Trust. Optimized library preparation workflows and high-performance sequencing platforms ensure strong mapping rates, reproducibility, and robust performance—even from low-input samples.
Comprehensive Analysis and Clear Reporting. Our bioinformatics pipeline delivers statistically rigorous analysis with publication-ready figures and transparent QC metrics.
Personalized Scientific Support. You work directly with experienced scientists from experimental design through final data review to ensure your study is properly powered and aligned with your biological objectives.
Active Motif’s Express RNA-Seq service includes:
- Total RNA isolation from cells. Additional fee for tissues
- RNA quality and integrity assessment
- Oligo-dT–primed reverse transcription
- Transposase-based library generation and QC
- Next-generation sequencing on the Illumina platform
- Comprehensive bioinformatics analysis: Raw data QC, Alignment to reference genome, Gene expression quantification, Differential expression analysis, Functional enrichment (GO), Data visualization (PCA plots, heatmaps, volcano plots)
- Multi-Omics Integration: Express RNA-Seq data can be integrated with additional epigenetic services such as: ChIP-Seq, ATAC-Seq, DNA methylation analysis. Through advanced bioinformatic integration, we help uncover mechanistic insights, linking chromatin state and transcriptional regulation.
Express RNA-Seq provides rapid, mRNA profiling without sacrificing data integrity. Whether you need fast results for a grant deadline, manuscript submission, or screening study, our streamlined workflow delivers actionable transcriptomic insights in just 4 weeks. Contact our expert scientists to get project-specific consultation to design the most appropriate experimental workflow.
Express RNA-Seq Workflow
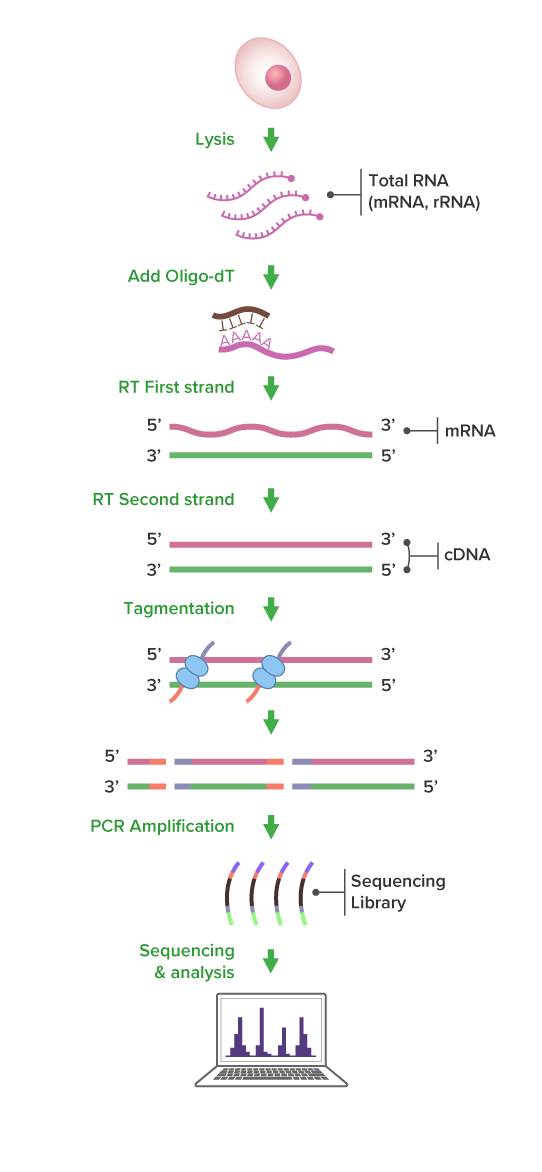
(Click image to enlarge)
Express RNA-Seq Services Data

(Click image to enlarge)
Figure 1. Performance Metrics are comparable between Express RNA-Seq and Illumina Stranded mRNA Prep.
Active Motif’s Express RNA-Seq exhibits similar rates of duplication and uniquely aligning reads compared to Illumina Stranded mRNA Prep, even at lower RNA input. Sequencing statistics are summarized in the Table below, showing that Express RNA-Seq has an increased percentage of exonic reads. Experiments were performed using THP-1 cells treated with LPS in triplicates, with 300 ng and 1,000 ng RNA input for Express RNA-Seq and 1,000 ng for Illumina Stranded mRNA Prep. Gene counts are reported for ~15 million reads, using PE50 for Express RNA-Seq and PE150 for Illumina Stranded mRNA Prep. Reads were aligned to the reference genome using STAR.

(Click images to enlarge)
Figure 2. Comparable gene detection between RNA-Seq methods.
Active Motif’s Express RNA-Seq detects a similar number of genes as the Illumina Stranded mRNA Prep, even at low RNA input. Experiments were performed using THP-1 cells treated with LPS in triplicates, with 300 ng and 1,000 ng RNA input for Express RNA-Seq and 1,000 ng for Illumina Stranded mRNA Prep. Gene counts are reported for ~15 million reads, using PE50 for Express RNA-Seq and PE150 for Illumina Stranded mRNA. Reads were aligned to the reference genome using STAR.

(Click image to enlarge)
Figure 3. High concordance of gene expression values between Express RNA-Seq and Illumina Stranded mRNA Prep.
Gene expression measurements obtained from Active Motif’s Express RNA-Seq show high correlation with those from Illumina Stranded mRNA Prep, indicating strong concordance between the methods. Experiments were performed using THP-1 cells treated with LPS in triplicates, with 1,000 ng RNA input for both workflows. Gene expression measurements are reported for ~15 million reads, using PE50 for Express RNA-Seq and PE150 for Illumina Stranded mRNA Prep. Reads were aligned to the reference genome using STAR.
 (Click image to enlarge)
(Click image to enlarge)Figure 4. Principal component analysis (PCA) of gene expression profiles shows minimal variance between Express RNA-Seq and Illumina Stranded mRNA Prep.
PCA of untreated and LPS-treated THP-1 cells demonstrates that the primary source of variance is LPS treatment (84%), with only 9% attributable to differences between the methods. Experiments were performed in triplicate using 1,000 ng RNA input per sample for Illumina Stranded mRNA prep and Express RNA-Seq. An additional set of Express RNA-Seq samples were performed with 300 ng input. Express RNA-seq samples were run in triplicate by two operators. Gene counts are reported for ~15 million reads using PE50 for Express RNA-Seq and PE150 for Illumina Stranded mRNA Prep. Reads were aligned to the reference genome using STAR.

(Click image to enlarge)
Figure 5. The identification of differentially expressed genes comparison is similar between Express RNA-Seq and Illumina Stranded mRNA Prep.
Volcano plot illustrating differential gene expression in LPS-treated THP-1 cells shows a similar number of up- and downregulated genes detected by Express RNA-Seq and Illumina Stranded mRNA Prep. Experiments were performed in triplicates using 1,000 ng RNA input per sample. Gene counts are reported for ~15 million reads, using PE50 for Express RNA-Seq and PE150 for Illumina Stranded mRNA Prep. Reads were aligned to the reference genome using STAR, and differentially expressed genes were identified using DESeq2.
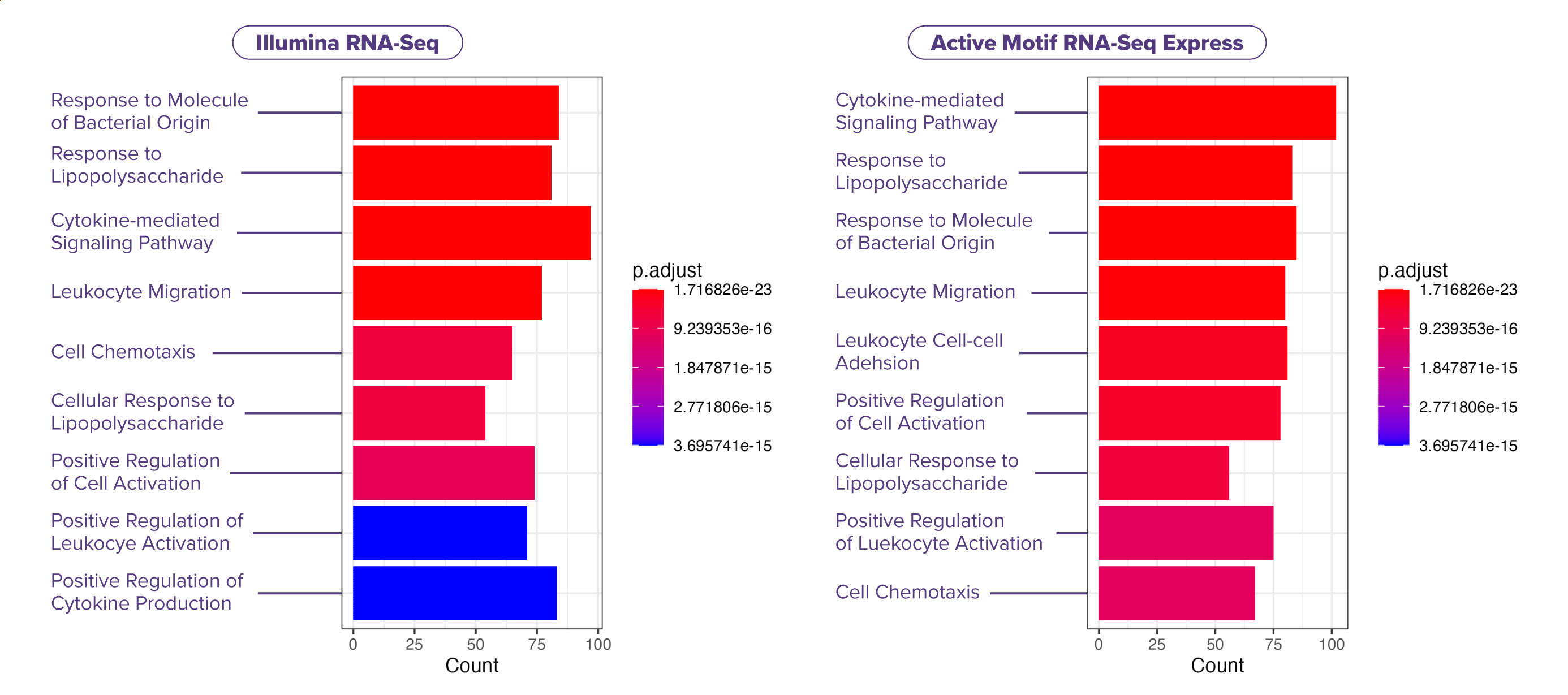 (Click image to enlarge)
(Click image to enlarge)Figure 6. Pathway analysis of upregulated genes identified similar LPS-response pathways by Express RNA-Seq and Illumina Stranded mRNA Prep.
Pathway analysis of LPS-treated THP-1 cells demonstrates that a similar subset of upregulated biological processes (GO terms) are detected by both Express RNA-Seq and Illumina Stranded mRNA Prep. Experiments were performed in triplicate using 1,000 ng RNA input per sample. Gene counts are reported for ~15 million reads, using PE50 for Express RNA-Seq and PE150 for Illumina Stranded mRNA Prep. Reads were aligned to the reference genome using STAR, and differentially expressed genes were identified using DESeq2.
Express RNA-Seq Services FAQs
1. Overview of Express RNA-Seq Services
Express RNA-Seq is a rapid, next-generation sequencing (NGS)–based RNA sequencing method designed for accurate and scalable gene expression profiling.
The workflow converts RNA into complementary DNA (cDNA), followed by a tagmentation-based library preparation step. During this step, transposase-mediated tagmentation simultaneously fragments and tags the cDNA with sequencing adapters in a single reaction. This streamlined approach reduces hands-on time, minimizes processing steps, and improves overall workflow efficiency.
Express RNA-Seq enables:
- Quantification of gene expression levels
- Detection of alternative splicing events
- Identification of novel transcripts
- Fusion gene discovery
- Differential gene expression analysis across biological conditions
This method is well suited for high-throughput transcriptomics studies, biomarker discovery, and comparative gene expression analysis.
Express RNA-Seq is optimized for speed and efficiency using tagmentation-based library prep, while standard Illumina mRNA-Seq focuses on maximal transcript coverage and depth.
- Illumina-based mRNA‑Seqis the standard workflow for sequencing polyadenylated transcripts. It provides genome-wide coverage, enabling detection of known and novel transcript isoforms, splice variants, and allele-specific expression, ideal for comprehensive transcriptome studies.
- Express RNA‑Seq is a rapid, streamlined sequencing service optimized for speed and efficiency. It includes a tagmentation-based library preparation step, which simultaneously fragments cDNA and adds sequencing adapters, reducing preparation time. Express RNA‑Seq uses efficient paired-end read configurations (like PE50) to deliver high-quality gene expression data quickly, making it ideal for projects that prioritize turnaround time without sacrificing essential transcriptome insights
Typical turnaround time is 4 weeks.
The exact timing depends on factors such as the number of samples, RNA quality, and desired sequencing depth.
2. Sample Requirements and RNA Quality
Express RNA-Seq input requirements depend on whether you are submitting purified RNA or cell pellets.
- Minimum input: 500 ng of total RNA / 20,000–50,000 mammalian cells / 20mg tissue
- Recommended input for optimal results: Up to 1 µg total RNA or 50,000–100,000 cells / 50mg tissue
Providing higher RNA input within this range improves library complexity, transcript detection sensitivity, and overall data robustness.
Before library preparation, we perform RNA quality control (QC), including: RIN and Qubit
| Input Type | Recommended Input | Minimum Requirement | Why Recommended? |
|---|---|---|---|
| Total RNA | Up to 1 µg | 500 ng | Improves library complexity, transcript detection sensitivity, and overall data robustness |
| Mammalian Cells | 50,000–100,000 cells | 25,000–50,000 cells | Provides sufficient RNA yield for optimal sequencing performance |
| Fresh Frozen Tissue | ≥50 mg | ≥20 mg | Ensures adequate RNA recovery and consistent library quality |
A RIN ≥ 7 is recommended for standard mRNA-Seq.
Lower RIN samples may be processed with customer approval.
Yes. RNA extraction is available for cell pellets and fresh frozen tissue.
Inquire here for all other sample types.
RNA samples should be shipped following the guidelines outlined in the Sample Preparation Form. Proper handling ensures sample integrity for accurate sequencing results.
3. Sequencing Depth and Run Configuration
We typically sequence 15–20 million reads per sample. This depth supports accurate gene expression profiling and differential expression analysis.
The optimal sequencing depth can vary depending on your experimental design, sample complexity, and goals, such as detecting low-abundance transcripts or performing transcript isoform analysis. Our team can help you determine the most effective read depth for your project to ensure high-quality results while minimizing sequencing costs.
Paired-end 50 bp (PE50) is the standard configuration, which provides reliable coverage and efficiency.
Paired-end 150 bp (PE150) is available for higher resolution applications.
4. Bioinformatics, Data Analysis and Deliverables
Our Express RNA-Seq bioinformatics pipeline typically includes:
- Raw data quality control (FastQC), assess read quality
- Adapter trimming, remove sequencing adapters
- Alignment to reference genome, map reads accurately
- Gene/transcript quantification, measure expression levels
- Differential expression analysis, identify genes with significant changes
- Functional enrichment analysis (GO), interpret biological significance
- Data visualization: PCA plots, heatmaps, and volcano plots
This comprehensive pipeline ensures that you receive high-quality processed data, meaningful insights, and ready-to-use visualizations for downstream research or publication.
Typical deliverables from an Express RNA-Seq project include:
- Raw FASTQ files, original sequencing reads
- Aligned BAM files, reads mapped to the reference genome
- Count matrices, gene expression quantification
- Differential expression tables, comparisons across conditions
- QC reports, quality control metrics for sequencing and alignment
- Bioinformatics summary report, comprehensive analysis and interpretation of results
These deliverables provide a complete dataset for downstream analysis and ensure you receive both raw and processed data, ready for further research or integration with other omics studies.
Yes. Express RNA-Seq services include publication-ready figures, such as:
- PCA plots, visualize sample clustering and variance
- Volcano plots, highlight differentially expressed genes
- Heatmaps, show expression patterns across samples
- Pathway enrichment plots, summarize biological pathway involvement
All figures are professionally formatted for publication and downstream interpretation, ensuring your RNA-Seq results are presentation-ready.
Yes. Express RNA-Seq data can be integrated with multiple omics datasets, including ChIP-Seq, ATAC-Seq, DNA methylation data, and CRISPR screening results. Integrating RNA-Seq with other omics data provides deeper mechanistic insights into gene regulation and biological pathways.
Express RNA-Seq Services Documents
Express RNA-Seq Services Sample Submission Portal
Our online sample submission portal allows you to easily upload your service project samples and track your project status. Follow the sample submission instructions in the portal to ensure that all your samples arrive at Active Motif in the best possible condition and properly associated with your project.
You might also be interested in:
| Name | Cat No. | Price | |
|---|---|---|---|
| Express RNA-Seq | 25218 | Get Quote |