<< Back to MOTIFvations Blog Home Page
Undruggable No More – Advances for Targeting Mutant Ras Proteins and Cancer
By Anne-Sophie Ay-Berthomieu, Ph.D.
May 17, 2022
Table of Contents:
Introduction
Ras is a small GTPase protein of 21kDa, seeming insignificant in size, but with a very unique destiny. Ras was first discovered in retroviruses where it was found to be involved in the oncogenic capability of RNA tumor viruses. Since then, homologs have been identified across prokaryote and eukaryote genomes.
Ras is activated by multiple signalling pathways and can, in turn, activate many downstream proteins and transcription factors. The broad Ras interactome means it is implicated in several developmental processes, as well as in pathological deregulation. In particular, Ras is widely involved in carcinogenesis and displays specific mutations which often correlate with distinct kinds of carcinoma(s) and with clinico-pathological features.
Until recently, Ras was thought to be “undruggable” because of the lack of structural pockets for drug interaction. However, with new findings from the protein family’s crystal structure, scientists have been able to create targeted compounds that bind to Ras protein, some of them even specific for certain mutants.
In this article, we will tell you the amazing story of Ras protein, how the worldwide effort of researchers was able to identify this protein family, decipher the complex signalling network regulated by Ras, and its involvement in cancer development. Ultimately, the goal is for the story to end with therapies which specifically target Ras.
Ras History: From Retrovirus Research to Cancer Research
Ras Discovery
Ras was identified from studies of transforming retroviruses isolated from rodents, cats, monkeys, chickens, and turkeys back in the 1960s. Harvey murine sarcoma virus (1964) and Kirsten murine sarcoma virus (1967) rapidly transformed animal cells in culture. Ras got its name from its ability to promote rat sarcoma and the different variants are named for each discoverer: H-Ras or Ha-Ras for Harvey and K-ras or Ki-ras for Kirsten. It was only in 1973 that Scolnick et al. hypothesized that the oncogenic property of these viruses resulted from the adoption of normal cellular sequences into their own genomes. Scolnick et al. wanted to confirm their hypothesis and led extensive, novel studies in the 70s-80s that identified the cellular origin of K-ras and H-ras. These genes code for a 21k Da protein with the ability to bind GDP and GDP and that associated with the plasma membrane. Unexpectedly, they found the cellular homolog of the viral gene and that this counterpart also binds to GTP/GDP and when overexpressed, where it induces cell transformation. These studies became the foundation of Ras biochemical and cellular analysis.
Unfortunately, these retroviruses were not originally recognized as associated with cancers and so research interest waned. However, finding that active eukaryotic DNA can be transferred into mammalian cells was used to develop a key technique – transfection by calcium phosphate. First experiments were performed in immortalized NIH/3T3 cell lines. After oncogene retroviral infection, NIH/3T3 displayed growth factor independence and lost contact inhibition, both hallmarks of malignancy. It was part of the beginning of “oncogene” characterization, including RAS and RAS mutations.
In 1982, three independent groups (Barbacid, Cooper, Weinberg) identified the same RAS gene as Kirsten and Harvey to be responsible for NIH/3T3 transformation in transfection assays. At the end of that same year, three other groups (Wigler, Barbacid, Chang) published the molecular basis of H-Ras activation in the EJ/T24 bladder carcinoma cell line. Unexpectedly, a single missense mutation in codon 12 was implicated in constitutive activation of H-Ras. This same mutation was also found in K-Ras and responsible for K-Ras activation in lung and colon tumor cells. In 1983, a third version of the RAS gene was discovered in neuroblastoma-derived DNA and called N-Ras. Later, the codon 12 mutation was found in human tumor tissue, but not in normal tissue, supporting the idea that Ras mutation was not simply a side-effect of in vitro cell passage. The realization that a single mutation can lead to carcinogenesis triggered a renewed, intense research area of cancer molecular mechanisms in the context of Ras and paved the way for the other Ras mutation identification.
Ras GTPase Protein Family
The RAS gene is highly conserved across the animal kingdom except for plants, where it’s not found. S. cerevisiae have 2 functionally redundant Ras proteins, Ras1 and Ras2, that are involved in spore viability. In Drosophila, Dras1 and Dras2 are RAS homologs found to play a role in eye development. The RAS gene encoding the LET-60 protein in C. elegans regulates vulval development. Finally, Ras proteins have also been found in Dictyostelium and zebrafish.
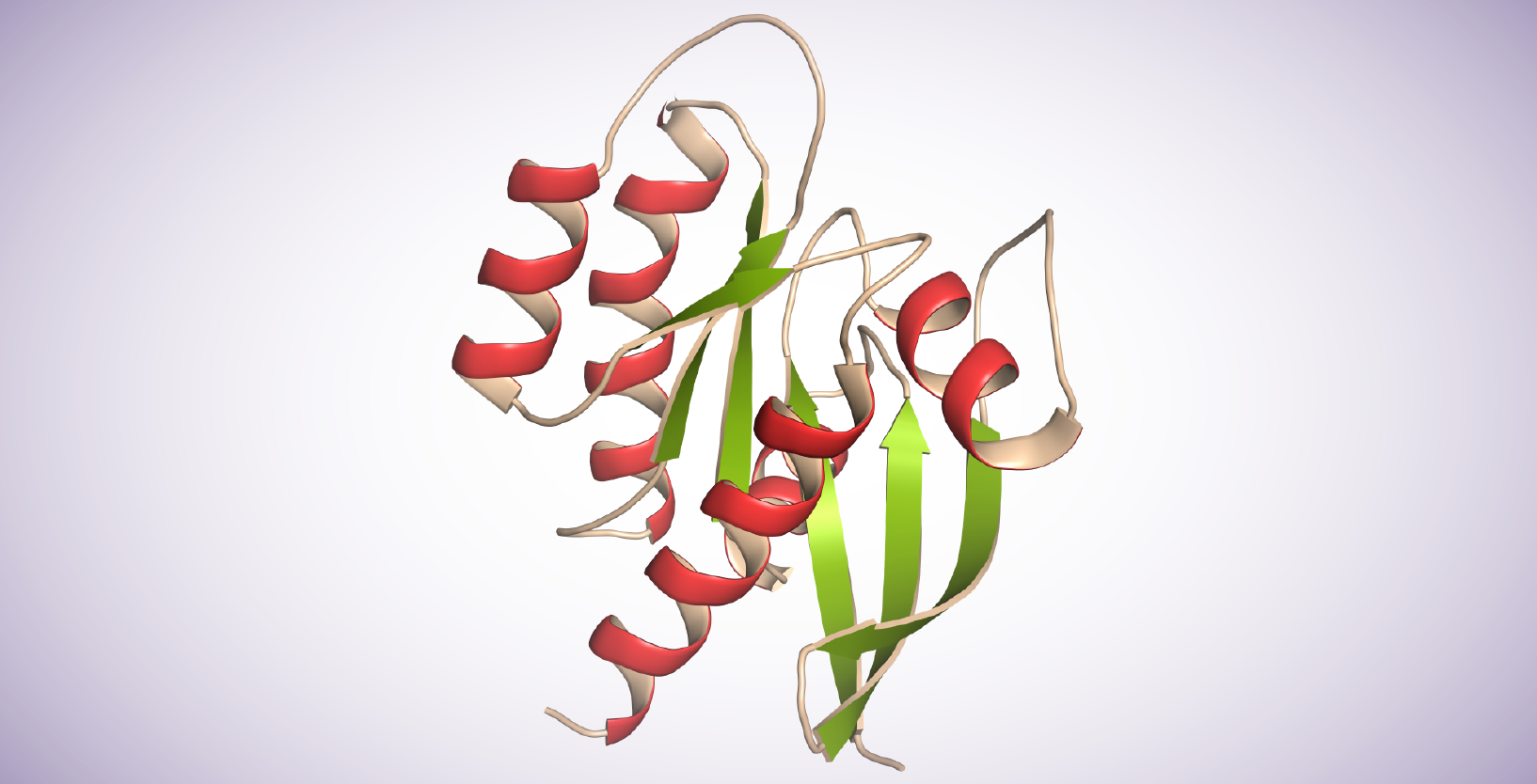
Sequence homology and structure analysis across species has been mapped into 5 branches in the RAS GTPase superfamily: Ras, Rho, Rab, Arf, and Ran. Ras proteins contain 6 beta-strands and 5 alpha helices. They possess a G domain and a C-terminal hypervariable region (HVRs). The G domain, containing switch I, switch II, and P loop sequences is responsible for GTP/GDP binding. The HVR domain is the target of post-translational modifications, including iso-prenylation, proteolysis, and methylation, mediating Ras-plasma membrane binding.
Ras proteins toggle between a GDP-bound inactive state and a GTP-bound active state in a GTP/GDP cycle which is tightly regulated. The switch between GDP and GTP is accelerated by Guanine nucleotide exchange factors (GEF), including SOS1, in response to upstream signalling. Conversely, GTPase activating proteins (GAPS) accelerate GTP hydrolysis. Ras activation induces signal transduction by binding to downstream effectors.
Ras localization is also particularly important and is activated only at the plasma membrane. This association necessitates Ras post-translational modifications: prenylation by the farnesyltransferase (FTase) or geranylgeranyltransferase (GGT), cleavage of the AAX terminal residues by Ras-converting enzyme (RCE1), and the methylation of the cysteine of the CAAX box by isoprenylcysteine carboxyl methyltransferase (ICMT). Once at the membrane, Ras undergoes oligomerization or dimerization.
Ras at the Center of a Complex Network
Ras is activated by different transmembrane receptors such as cytokine receptors, receptor tyrosine kinases (RTKs), G-protein coupled receptors (GPCRs), and extracellular matrix receptors. It is subsequently able to transduce the signal to various downstream effectors containing a Ras-binding domain (RBD), regulating transcription in a tissue-type and microenvironment specific manner.
Ras: a Major Driver of Carcinogenesis
RAS Mutations
Ras is mutated in 19% of all cancers, which is why it’s been a center of attention in the cancer research field. The current dogma is that K-Ras is the most frequently mutated in cancer, followed by N-Ras and H-Ras.
Pancreatic Ductal Adenocarcinoma (PDAC), Lung Adenocarcinoma (LUAC), Colon, and Rectal adenocarcinoma carry K-Ras mutations at a frequency of 66.1%, 16.5%, 30.3%, and 34.4% respectively. Conversely, in hematopoietic cancers such as chronic myelomonocytic leukemia (CMML) and acute myeloid leukemia (AML), N-Ras mutations occur at 13.1% and 13.6% frequency. In melanoma, thyroid carcinoma, and larynx carcinoma at N-Ras mutations occur at 18.6%, 8.1%, and 9.7% frequency. H-Ras mutations are quite rare in human carcinoma but can be found in salivary gland, mouth, and vulvar carcinoma.
Over 100 mutations have been detected in RAS genes, but G12, G13, and Q61 remain the dominant “hot spots” of mutation in K-Ras and N-Ras. About 80% of K-Ras mutations are located at G12, whereas 60% are at the Q61 locus for N-Ras. Why some mutations are more frequent in specific conditions is still unknown. However, Ras mutations confer several characteristics to tumors: increased proliferation abilities, promoted epithelial to mesenchymal transition, and overall acceleration of tumor development.
A more thorough review can be found in the Catalog Of Somatic Mutations in Cancer (COSMIC).
Ras: a Diagnostic and Prognostic Biomarker
Studies of cancer patient cohorts and Ras mutations have highlighted correlations between Ras mutations and clinico-pathological features (review here). A very striking example is in colorectal cancer (CRC). Patients bearing K-Ras or N-Ras mutations have shorter overall survival rates. Moreover, K-Ras mutations are strongly correlated with metastasis spreading to the lung instead of the liver. In melanoma, N-Ras mutations interfere with mitoses and inhibit tumor-infiltrated lymphocytes. K-Ras mutations in ovarian cancer correlate with lower differentiation and higher progesterone expression.
Besides being a prognostic tool, Ras mutational status can also indicate therapeutic efficacy. In metastasis of CRC, K-Ras mutations predict resistance to EGFR inhibitor therapy (e.g., cetuximab). Ras mutations also seem to forecast the efficiency of therapy targeting negative regulators such as the immune checkpoint blockade (ICB). Indeed, the protein PD-L1, the target of the ICB, correlates with K-Ras mutations in pulmonary sarcomatous carcinoma and lung adenocarcinoma, suggesting a better ICB efficiency.

Ras: a Druggable Therapeutic Target
Given the high frequency of Ras mutations in cancer, developing targeted therapies would be an important advance for treatment. However, Ras is also important for normal physiological processes. That being the case, drugs need to target Ras mutants specifically to avoid deleterious secondary effects.
K-RasG12C Targeting Therapy
As mentioned earlier, Ras was thought to be undruggable because of the lack of potential inhibitor-binding regions and its picomolar affinity for GTP/GDP. However, in 2013, Kevan Shokat’s team successfully reported the development of compounds that irreversibly bound to K-RasG12C in the GDP-bound state, blocking SOS activation and RAF association. These compounds relied on the mutant cysteine for binding and so did not affect wild-type protein. In addition, crystallographic studies revealed the formation of a binding pocket not seen earlier in earlier structures. Binding of the inhibitors to K-RasG12C disrupted the binding pocket and subsequently shifted the affinity of K-Ras to prefer GDP over GTP. These discoveries paved the way for renewed drug discovery efforts which targeted mutant Ras.
Amgen was the first to successfully bring one of the many resulting new K-Ras inhibitors to market. In May 2021, the US Food and Drug Administration approved K-RasG12C inhibitor Lumakras (sotorasib: AMG510) for previously treated patients with metastatic NSCLCC expressing the K-RasG12C mutation. It was reported in April 2022 that patients treated with sotorasib had a two-year overall survival rate of 32.5 percent, an improvement over previous therapies. Long-term treatment with sotorasib was well tolerated, with mild and manageable toxicities. However, most patients do not respond, due to intrinsic or acquired resistance to the drug.
Another K-RasG12C inhibitor, adagrasib: MRTX849, from Mirati may soon be available to patients. This inhibitor was chemically derived from AMG510 and is similar in structure. A drug application for Adagrasib was accepted by the USFDA in February 2022, for treatment of patients with previously treated K-RasG12C NSCLC. Again, drug resistance is an ongoing issue in patients. However, new therapeutic strategies are being tested, for example combining K-RasG12C inhibitors with other compounds to increase efficacy.
In addition to the inhibitors above, there are many other K-RasG12C inhibitors in development by both Amgen and Mirati and by other pharmaceutical companies such as Johnson & Johnson, LOXO Oncology, Boehringer Ingelheim, Revolution Medicines, Sanofi, and others.
Other Mutants
For other K-Ras G12 mutants, such as G12D and G12V, a different strategy is necessary because aspartic acid and valine are less reactive than cysteine. Also, unlike K-RasG12C these mutants do not spend as much time in the GDP state. A 2022 Nature paper by Shokat describes how these other mutants can still be inhibited by noncovalent ligands, and that the inhibition does not necessarily depend on the GDP state of K-Ras. The results point to new therapeutic opportunities for cancers driven by these other mutations. One of these K-RasG12D inhibitors, Mirati’s MRXT1133 is already showing clear tumor regression in preclinical pancreatic cancer models.
Another approach has been to target all mutations and develop a “pan-Ras” inhibitor. One such compound, compound 3144 is very efficient against tumor proliferation and survival without significant adverse effects. It binds K-RasG12D, wild-type K-Ras, N-Ras, and H-Ras at D38, A59, and Y32.
Finally, PROteolysis TArgeting Chimeras (PROTACs), which are small molecules capable of removing specific unwanted proteins, are being explored as a way to stop mutant Ras. PROTACs make use of the cell's ubiquitin–proteasome system (UPS) by bringing together the target protein and an E3 ligase, enabling it to be ubiquitinylated and degraded. One such PROTAC is LC-2, a compound which is selectively capable of inducing K-RasG12C protein degradation.
Cancer Vaccines
This approach aims to induce an immunological response against specific Ras protein mutants. Peptides from mutant Ras proteins are injected into the patient in combination with recombinant GM-CSF to activate dendritic cells and enhance the T cell response against these specific mutations. Targovax’s therapeutic anti-cancer vaccine, TG01 is currently being tested for pancreatic cancer. TG01 injection in PDAC patients shows an increase in immune response and overall survival. Another drug, TG02, is being tested in CRC patients.
An mRNA vaccine developed by Moderna, mRNA-5671, is also in a clinical trial. It codes for K-Ras mutations and is formulated in lipid nanoparticles. It is used as a single agent or in combination with pembrolizumab.
Indirect Approach - Inhibitor of Nucleotide Exchange
GTPase activity is crucial for Ras function. Thus, inhibiting GDP-GTP exchange can also block Ras activity. SOS proteins are guanosine nucleotide exchange factors (GEF) that increase the Ras nucleotide exchange rate of GDP for GTP. So, a different approach is to develop small molecules that bind directly to SOS rather than Ras. Among these is BAY-293, which inhibits the Ras-SOS1 interaction. Used alone, it has weak physiological effects on K-Ras mutant proteins. However, in combination with ARS-853, a K-RasG12C inhibitor, BAY-293 shows a synergistic growth-inhibitory effect, suggesting that this kind of small molecule could be used in combination for effective treatment. Another compound, BI-1701963 is being tested both alone and in combination with trametinib (inhibitor of MEK) for patients with solid tumors and Ras mutations.
SHP2 Inhibitors
SHP2 is another protein which promotes Ras nucleotide exchange. The goal of targeting SHP2 is the same as SOS inhibitors: inhibiting nucleotide exchange and therefore, Ras GTPase activity. Revolution Medicines and Sanofi are co-developing a compound called RMC-4630, a potent SHP2 inhibitor in a multi-cohort Phase 1/2 clinical program. RMC-4630, is currently in trials as both a monotherapy and in combination with comibetinib.
Inhibitor of Ras Processing
This strategy aims to prevent Ras localization to the plasma membrane. Ras undergoes several steps of post-translational modification before attaching to the membrane, including isoprenylation. Farnesyltransferase (FTase) is an enzyme that facilitates isoprenylation of Ras. While in K- and N-Ras there are redundant methods for isoprenylation, H-Ras can only be prenylated by FTase. Therefore, FTase inhibitors such as Tipifarnib are being studied for treatment of cancers involving H-Ras mutations.
ICMT, is another enzyme involved in post-translational modification of Ras and therefore localization of Ras to the plasma membrane. A ICMT inhibitor, cysmethynil, slows down Ras-mutant cells proliferation. Another ICMT inhibitor, UCM-1336 prevents Ras-membrane association in all four isoforms, regardless of the mutation, and reduces cell proliferation. None of these are in a clinical trial yet and need enhancement for therapeutical use as these enzymes also process other substrates and blocking their action could lead to important off-target effects.
Summary
The Ras family is at the center of several crucial physiological processes across species and plays an important role in tumorigenesis, including CRC, NSCLCC, and PDAC. It is at the center of a complex network, regulating different transcriptional programs. Volumes of work have been done on Ras and the story continues today - to decipher Ras function, and to pursue understanding of Ras mutations for diagnostic and prognostic tools.
Ras mutations are now well described in the literature and are used as diagnostic and prognostic tools. Ras mutations are also able to predict the efficiency of certain therapies.
All these characteristics lead scientists closer to new discoveries for ways to target Ras protein and its mutants as well as to characterize Ras activity. Several clinal trials are currently running with already pre-clinical encouraging results. Being able to efficiently target Ras mutants paves the way to precision medicine.
About the author

Anne-Sophie Ay-Berthomieu, Ph.D.
Anne-Sophie was born in the south of France and grew up between the Mediterranean Sea and the Pyrenean Mountains. She grew up as a science fiction fan, leading her to specialize in molecular biology and genetics during graduate school at the University of Lyon, France (secretly hoping her research would give her superpowers!). After living in different places for work, she is back in Lyon, France where she shares her time between her husband, her family, and her friends. During her free time, Anne-Sophie challenges herself with hiking, climbing, racing, and traveling in foreign countries – while waiting for her superpowers to grow!
Contact Anne-Sophie on LinkedIn with any questions, or to tell her about your superpowers.
Related Articles
Oncohistones: Histone Mutations & Their Oncogenic Effects
June 24, 2021
Oncohistones are amino acid mutations in Nucleosomes shown to have strong correlation to many types of cancers. The cancer-type specificity of oncohistones may facilitate targeted therapy, while limiting the side effects that occur with chemotherapy. Methods to detect these mutations could also help in diagnostic and prognostic applications in personalized medicine.
Read More
PROTAC-mediated Targeted Protein Degradation in Cancer: PARP, EGFR, and SMARCAs in Focus
April 13, 2022
Proteolysis-Targeting Chimera (PROTAC) protein degraders are the emerging alternative to small molecule-based targeting. Here we look at key protein targets and discuss how PROTAC-mediated targeted protein degradation represents a promising new approach to cancer treatment.
Read More
<< Back to MOTIFvations Blog Home Page



