<< Back to MOTIFvations Blog Home Page
3’-Digital Gene Expression (3’-DGE) Sequencing: High Through-put at Low Cost
June 14, 2022
Table of Contents:
Introduction
Over the past decade, RNA sequencing (RNA-Seq) has been a highly effective technique for delivering high resolution information on the transcriptome, the set of all RNA transcripts in either an individual cell or population of cells. Transcriptome data can provide deep insight into a whole host of processes, ranging from transcriptional regulation and cell differentiation to carcinogenesis.
Successful RNA-Seq starts with high-quality library preparation from RNA. While there have been various workflows developed for library construction, it typically involves fragmenting the RNA, converting the RNA to complementary DNA (cDNA), adapter addition, and amplification of the library to sufficient quantity for next-generation sequencing. There have been successful attempts at reducing the time and cost for these library prep steps. However, the sequencing depth requirements to determine the transcripts remain the same, typically 20-30 million or more paired end reads. Sequencing costs have fallen over time. But it’s still quite expensive, especially when working with hundreds of samples.
There is a lesser-known alternative method of library prep, which considerably reduces sequencing depth requirements without sacrificing high quality gene expression profiling data. Known as 3'-Digital Gene Expression (3'-DGE), this method requires only 3-10 million single end reads. While it is not suitable for all RNA-seq applications, when used appropriately, the result is differential expression analysis and quantitative transcriptome profiling at a much lower cost.
What is 3'-Digital Gene Expression (3'-DGE) RNA-Seq?
There are many applications for high-throughput bulk RNA-Seq including transcriptome assembly, characterization of alternative splice sites or transcript isoforms, variant detection, and profiling allele-specific expression. The primary goal of a typical RNA-Seq project, however, is to identify genes and pathways that are up- or down-regulated in response to some experimental condition.
While there are many techniques for performing RNA-seq experiments, 3'-Digital Gene Expression (3'-DGE) is a relatively new mode of RNA sequencing that targets the 3'-end of each transcript and is optimized for such differential gene expression projects. The "digital" in 3'-Digital Gene Expression refers to counting and this approach to RNA sequencing is sometimes called a "transcript counting" method.
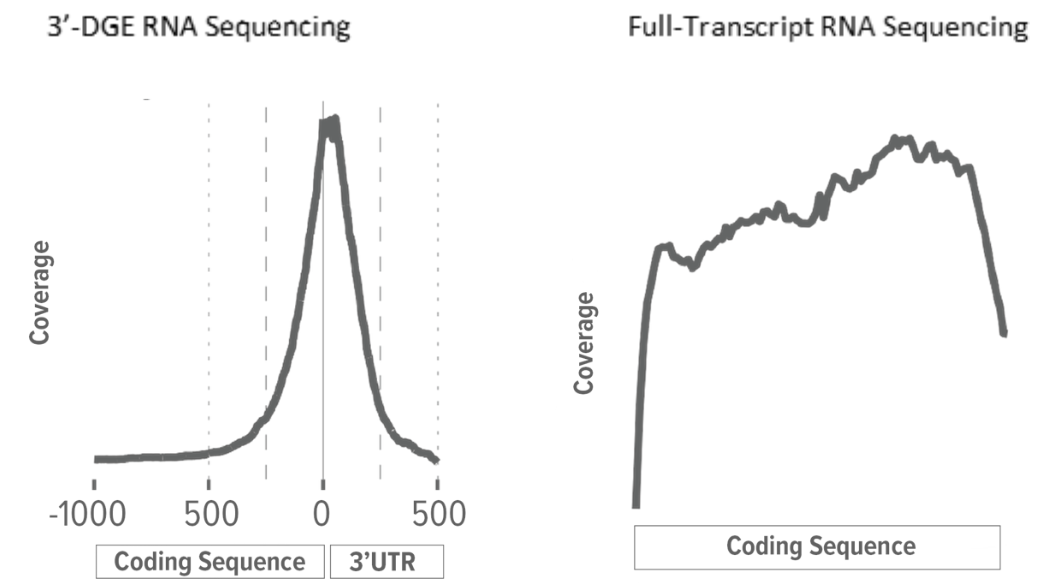
How does 3'-DGE compare to full transcript RNA sequencing?
Conventional full-transcript (FT) coverage RNA-Seq yields sequencing reads that map to the entire expressed transcriptome. This is achieved through the combination of random RNA fragmentation and subsequent random priming during the cDNA synthesis step. The result is sequencing reads that, when mapped to the reference transcriptome or genome, tile across the exons of expressed transcripts. With conventional RNA-Seq, the number of reads mapping to a gene is a function of transcript abundance and transcript length. A long gene, therefore, will have more reads mapping to it than a short gene of similar transcript abundance.
Rather than use random priming, the cDNA synthesis step for 3'-DGE RNA-Seq libraries uses an adapter with an oligo(dT) to prime off the transcript's poly(A) tail. This means that 3'-DGE reads tend to map only to the 3'-end of the transcript. Since there is a theoretical 1-to-1 correlation between transcript molecule and cDNA molecule, transcript length is irrelevant and the number of reads mapping to a gene is solely a function of transcript abundance. A long gene, therefore, will have the same number of reads mapping to it as will a short gene of similar transcript abundance.

Advantages & Limitations of 3'-DGE RNA-Seq
There are three significant limitations to the use of 3'-DGE RNA-Seq. Most importantly, 3'-DGE is limited to eukaryotic organisms since the first-strand synthesis relies on priming off the poly(A) tails of transcripts. Since the majority of 3'-DGE reads can map to the 3'-UTR of a gene, it is also best to have a well-annotated transcriptome. There is a workaround for this, however. For species with known translation stop sites, but no annotated transcription stop sites, it’s possible to bioinformatically create a reference sequence or annotation with regions of interest based on some distances upstream and downstream from the translation stop site. To avoid issues related to over-estimating 3'-UTR lengths, this workaround works best with strand-specific RNA sequencing to distinguish reads mapping to a nearby 3'-UTR of an adjacent gene on the opposite strand. The third limitation concerns analysis flexibility. Researchers performing analyses that require data from the entire transcript (assembly, splicing, etc.) will want to stick with conventional RNA-Seq or use a strategic combination of the two approaches. With those limitations in mind, perhaps the primary reason more researchers are not using 3'-DGE as their RNA-Seq of choice is simply that the method and its advantages are not well known.
The 1-to-1 correlation between transcript molecule and cDNA molecule is what makes 3'-DGE an effective transcript counting method, reducing the complexity of data analysis. To illustrate this point, consider an RNA-Seq sample with 100 reads mapping to Gene A. With conventional RNA-Seq, it is unclear how many unique transcript molecules (is it 1?, 10?, 25?, or ?) gave rise to these 100 reads. This uncertainty is true even with transcript-length normalization. Those 100 reads in a 3'-DGE experiment, however, essentially correspond to 100 transcript molecules.
This reduced complexity associated with 3'-DGE confers one of the method's biggest advantages. Transcript counting means that less sequencing depth is needed for a robust analysis compared to conventional RNA-Seq. Also, paired-end sequencing does not add value for 3'-DGE. So, a researcher who might sequence their conventional RNA-Seq libraries at 20-30 million pairs of paired-end reads per sample could significantly stretch their research budget by sequencing those same samples as 3'-DGE libraries at 3-10 million single reads per sample. Due to budgetary constraints, researchers are often forced to make compromises on replication in RNA-Seq experiments. The economical yet robust 3'-DGE alternative eases these constraints and allows for increased replication and, therefore, increased statistical power for more accurate differential gene expression analyses.

Discoveries enabled by using 3'-DGE
RNA can be scary to work with. The scourge of lurking RNases has caused countless restless nights for researchers planning their first (or hundredth!) RNA extraction. Seeing post-RNA-extraction QC results with high RIN (RNA Integrity Number) scores brings great relief. It is not uncommon, however, for a project to have variability in RNA quality, especially as sample sizes grow. While this can cause significant issues for conventional RNA-Seq libraries that rely on the presence of the full transcript, 3'-DGE libraries are less sensitive to such variability. This advantage can be taken a step further. It is impossible to get high-quality RNA from some sample types regardless of the RNA extraction method. In situations like this, 3'-DGE has the potential to give good differential gene expression results where conventional RNA-Seq might fail.
Variant detection has the potential to add a new dimension to a comprehensive RNA-Seq analysis. This is especially true when sequencing a population of individuals. Given that protein-coding exons are under greater selective pressure than are non-coding regions of the gene, 3'-DGE reads should be enriched for variants compared to reads from conventional RNA-Seq. This additional variant information can be useful for analyses beyond gene expression studies, including characterizing BILs/NILs (backcross inbred lines/near isogenic lines) and performing GWAS or mapping QTL. A recent study took advantage of this reduced selective pressure and used 3'-UTRs to finally resolve the avian family-level tree of life (view the publication).
We highly recommend considering 3’DGE for any RNA-seq project involving a well annotated eukaryotic genome and where transcript-splicing information is not required. This approach delivers numerous advantages, including half the sequencing cost, easier data analysis, and less sensitivity to RNA quality. Get started with 3’DGE sequencing using one of Active Motif’s YourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep Kits.
About the author

Mike Covington
Mike was co-founder and CTO of Amaryllis Nucleics, an RNA-Seq company that recently joined Active Motif. Before transitioning to bioinformatics and next-gen sequencing, Mike started his career at the bench as a molecular biologist studying the physiological relevance of the plant circadian clock. As an open-source science and software enthusiast, he has spent the last decade focused on developing bioinformatic tools, science-related web apps, and new molecular biology tools. A native of the Pacific Northwest, Mike has spent most of his life up and down the West Coast where he enjoys adventures in/on the water, experimenting with music and languages, reading with a cat on his lap, and creating vegan junk foods.
Related Articles
Library QC for ATAC-Seq and CUT&Tag AKA “Does My Library Look Okay?”
December 8, 2021
“Does my library look okay?” is the most common question posed to Active Motif technical support. Get the practical scoop on quality control for ATAC-seq and CUT&Tag libraries. Find out how much library to expect from different libraries, what they should look like, and what to do if it’s not as expected.
Read More
PROTAC-mediated Targeted Protein Degradation in Cancer: PARP, EGFR, and SMARCAs in Focus
April 13, 2022
Proteolysis-Targeting Chimera (PROTAC) protein degraders are the emerging alternative to small molecule-based targeting. Here we look at key protein targets and discuss how PROTAC-mediated targeted protein degradation represents a promising new approach to cancer treatment.
Read More
<< Back to MOTIFvations Blog Home Page



