YourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep Overview
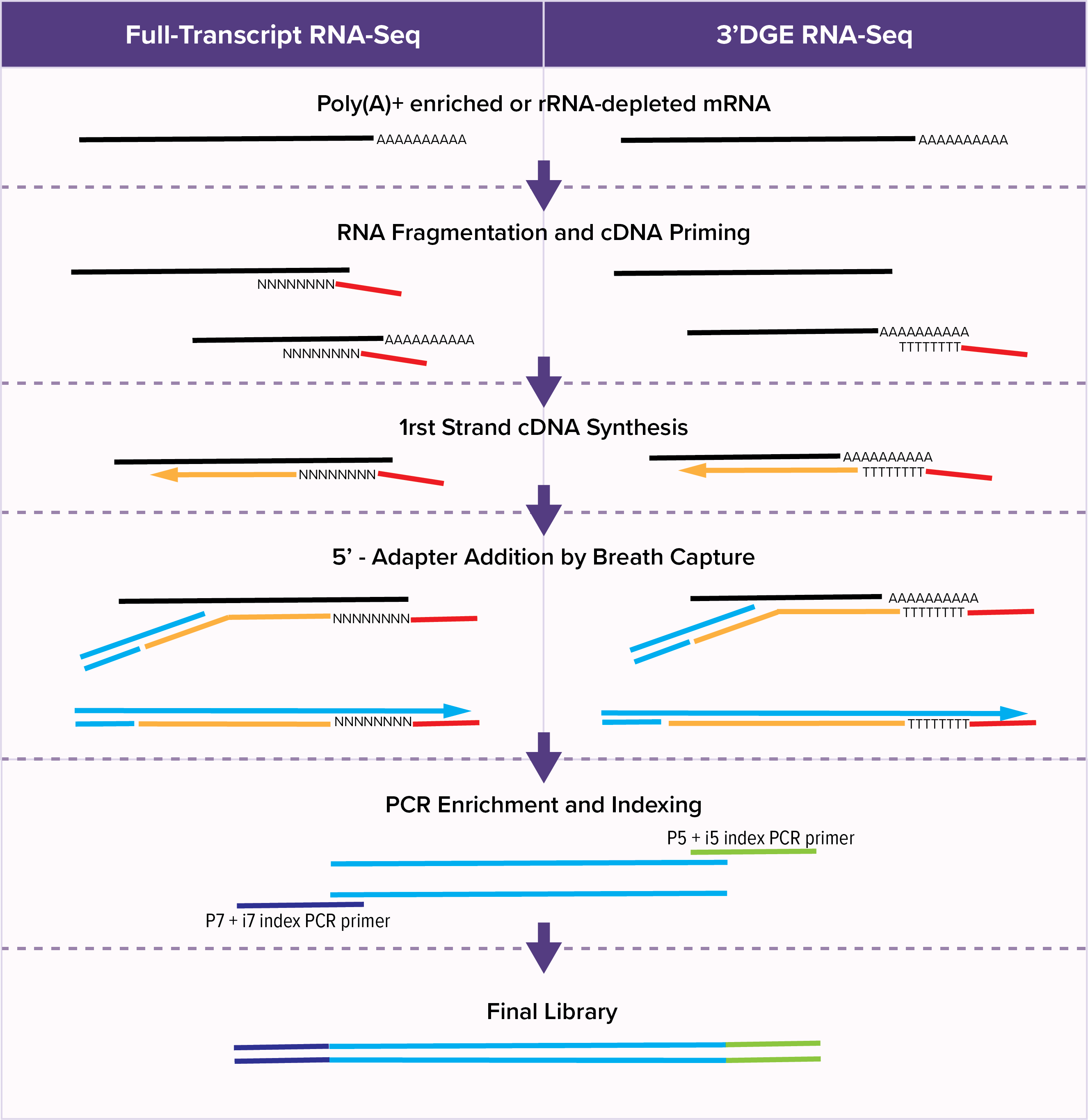
Active Motif’s YourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep Kits provide a fast and easy method for either conventional Full-Transcript (FT) or 3’-Digital Gene Expression (DGE) sequencing.
Conventional full-transcript (FT) RNA-seq yields sequencing reads that map to the entire expressed transcriptome. The 3’ Digital Gene Expression (3’-DGE) option creates reads mapping just at the 3’-end of the transcript. Because of the reduced complexity, 3’-DGE requires a quarter to a third less sequencing depth, greatly reducing costs.
The YourSeq Library Prep workflow takes advantage of the natural phenomenon of DNA breathing to eliminate several steps which add time and cost to traditional RNA library prep protocols. DNA breathing is the opening and closing of DNA due to thermal fluctuations. Following first-strand cDNA synthesis, the YourSeq Library Prep adds the 5-prime adapter to the cDNA when the terminal end of the DNA:RNA hybrid molecule is open in a process called “Breath Capture.” This allows the enzymatic synthesis of a second strand of DNA without a step requiring the removal of RNA. This workflow also implements strictly enforced strand specificity, since the adapter is added by priming off the cDNA strand and cannot prime off the RNA strand.
YourSeq (FT & 3’DGE) Strand Specific Library Prep Kits can be ordered with either combinatorial dual indexes or unique dual indexes. Combinatorial dual-indexing (CDI) kits allow you to choose from 96 or 384 unique index combinations. Unique dual-indexing (UDI) kits allow the multiplexing of either 24 or 96 samples at a time without using any index twice. This can help avoid Illumina index hopping, or the incorrect assignment of libraries to the expected index.
 (Click image to enlarge)
(Click image to enlarge)YourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep Highlights:
- Only 4 hours from RNA to library prep
- Option for both full-transcript (FT) or 3’-Digital Gene Expression (DGE) sequencing
- Strictly enforced strand specificity
- Available with Combinatorial Dual Indexing (CDI) or Unique Dual Indexing (UDI)
FT vs. 3’DGE
The YourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep Kits come with sufficient reagents for two types of RNAseq libraries, full-transcript (FT) or 3’-Digital Gene Expression (3’DGE). There are advantages and disadvantages to both methods. Use the appropriate workflow for your needs.
3’ Digital Gene Expression (3’DGE) Sequencing
The 3’ Digital Gene Expression (3’-DGE) option is optimized for differential gene expression analyses, where the main goal is to identify genes and pathways that are up- or down- regulated in response to some experimental condition.
Rather than use random priming, the cDNA synthesis step for 3’-DGE RNA-seq libraries uses an adapter with an oligo(dT) to prime off the transcript’s poly(A) tail. This means that 3’-DGE reads tend to map only to the 3’-end of the transcript. Since there is a theoretical 1-to-1 correlation between transcript molecule and cDNA molecule, transcript length is irrelevant and the number of reads mapping to a gene is solely a function of transcript abundance. A long gene, therefore, will have the same number of reads mapping to it as will a short gene of similar transcript abundance.
The reduced complexity associated with 3’-DGE confers one of the biggest advantages. Transcript counting means that less sequencing depth is needed for a robust analysis compared to conventional RNA-seq. Also, paired-end sequencing does not add value for 3’-DGE. So, a researcher who might sequence conventional RNA-seq libraries at 20-30 million pairs of paired-end reads per sample can reduce cost by sequencing those same samples as 3’-DGE libraries at 3-10 million single reads per sample.
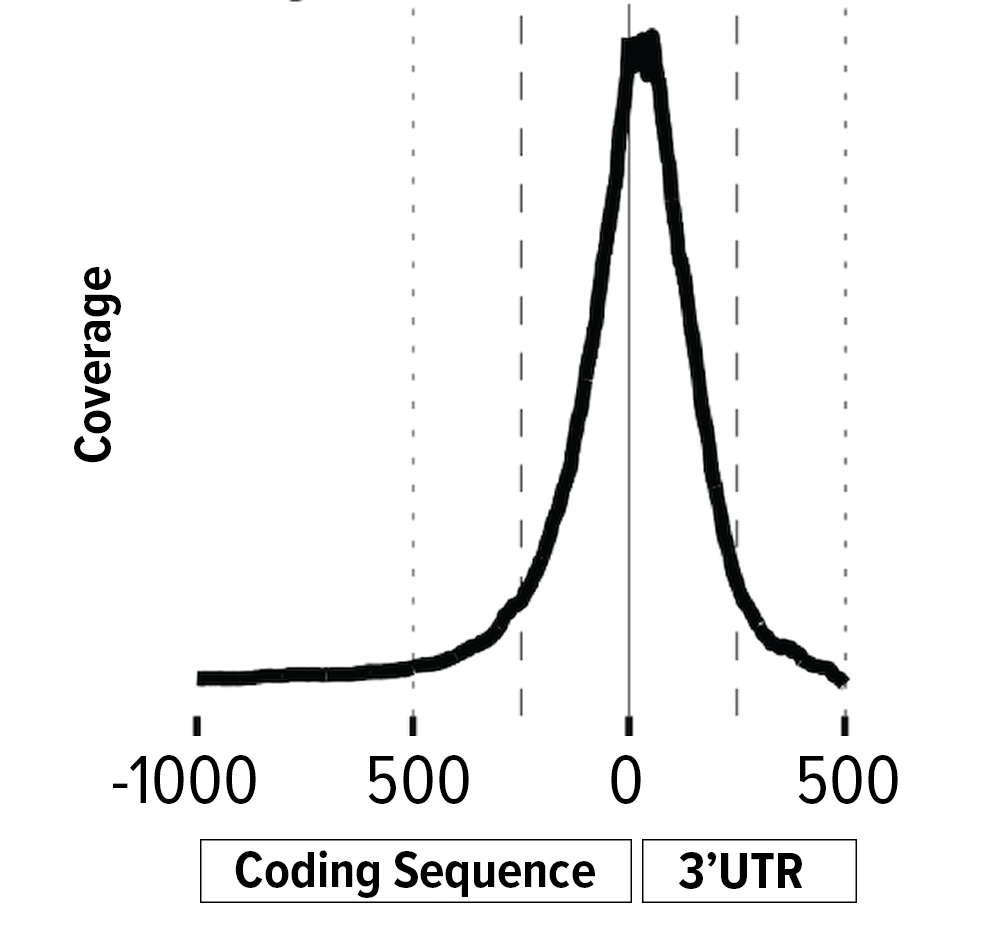
3’DGE RNA sequencing is optimal for:
- Differential gene expression for well-annotated genomes
- To improve annotation by identifying expressed genes and their 3’-UTRs
- Genotyping and detection of polymorphisms in the 3’-UTR
- Detection and characterization of alternative polyadenylation
Full Transcript (FT) Sequencing
The Full Transcript (FT) Sequencing option is for conventional full transcript RNA sequencing. This is the most common type of RNA sequencing that people are familiar with. Conventional full-transcript (FT) coverage RNA-seq yields sequencing reads that map to the entire expressed transcriptome. This is achieved through the combination of random RNA fragmentation and subsequent random priming during the cDNA synthesis step. The result is sequencing reads that, when mapped to the reference transcriptome or genome, tile across the exons of expressed transcripts. With conventional RNA-seq, the number of reads mapping to a gene is a function of transcript abundance and transcript length. A long gene, therefore, will have more reads mapping to it than a short gene of similar transcript abundance.
FT sequencing requires 20-30 million paired-end reads per sample, while 3’-DGE libraries require only 3-10 million single reads. However, there are circumstances where full-transcript sequencing is required. For example, splice variant analysis or the detection of exonic polymorphisms require reads from the entire coding sequence.
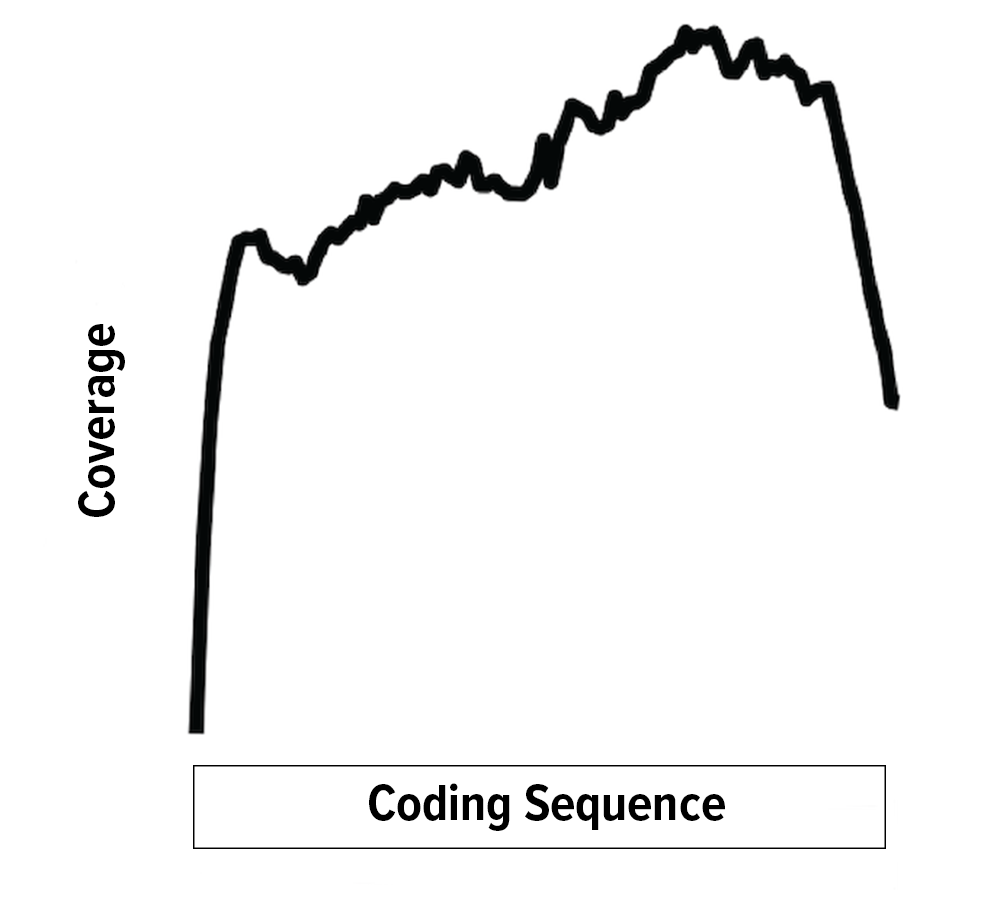
Full-transcript RNA sequencing is optimal for:
- Differential gene expression for non-annotated genomes
- Genotyping and detection of exonic polymorphisms
- Splice variant analysis
- Detection of gene fusions abundance
YourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep Contents
All components should be stored at -20°C until first use. Once Carboxyl Beads have thawed, store at 4°C.
| Catalog no. | 23001 | 23002 | 23003 | 23004 |
|---|---|---|---|---|
| Indexing | CDI | CDI | UDI | UDI |
| Reactions | 24 | 96 | 24 | 96 |
| Reagent | ||||
| FT Priming Mix | 75 μl | 300 μl | 75 μl | 300 μl |
| DGE Priming Mix | 75 μl | 300 μl | 75 μl | 300 μl |
| Control mRNA | 5 μl | 5 μl | 5 μl | 5 μl |
| First Strand Mix | 135 μl | 540 μl | 135 μl | 540 μl |
| RT enzyme | 15 μl | 60 μl | 15 μl | 60 μl |
| Breath Capture Mix | 175 μl | 690 μl | 175 μl | 690 μl |
| 5-Prime Adapter | 120 μl | 480 μl | 120 μl | 480 μl |
| DNA Pol I | 10 μl | 30 μl | 10 μl | 30 μl |
| PCR Mix | 264 μl | 1056 μl | 264 μl | 1056 μl |
| Phusion Pol | 8 μl | 24 μl | 8 μl | 24 μl |
| Nuclease-Free Water | 1.8 ml | 1.8 ml | 1.8 ml | 1.8 ml |
| 10 mM Tris HCl | 1.8 ml | 5 ml | 1.8 ml | 5 ml |
| Carboxyl Beads Buffer | 1.8 ml | 6 ml | 1.8 ml | 6 ml |
| Carboxyl Beads | 1.5 ml | 6 ml | 1.5 ml | 6 ml |
| YourSeq Index i5-A | 30 μl | 120 μl | ||
| YourSeq Index i5-B | 30 μl | 120 μl | ||
| YourSeq Index i5-C | 30 μl | 120 μl | ||
| YourSeq Index i5-D | 30 μl | 120 μl | ||
| YourSeq i7 1-24 Index Set | 1 plate | 1 plate | ||
| YourSeq i5 1-24 Index Set | 1 plate | |||
| YourSeq i7 1-96 Index Set | 1 plate | 1 plate | ||
| YourSeq i5 1-96 Index Set | 1 plate | |||
YourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep FAQs
What are the advantages of the YourSeq RNA-seq workflow?
The YourSeq Library Prep workflow takes advantage of the natural phenomenon of DNA breathing to eliminate several steps which add time and cost to traditional RNA library prep protocols. DNA breathing is the opening and closing of DNA due to thermal fluctuations. Following first-strand cDNA synthesis, the YourSeq Library Prep adds the 5-prime adapter to the cDNA when the terminal end of the DNA:RNA hybrid molecule is open in a process called “Breath Capture.” This allows the enzymatic synthesis of a second strand of DNA without a step requiring the removal of RNA. This workflow also implements strictly enforced strand specificity, since the adapter is added by priming off the cDNA strand and cannot prime off the RNA strand.
When do I choose Combinatorial Dual Indexing (CDI) versus Unique Dual Indexing (UDI)?
Combinatorial Dual Indexing (CDI) uses a unique combination of Index 1 (i7) and Index 2 (i5) indexes for each sample. However, samples may share index sequences. Unique Dual Indexing (UDI) uses a unique combination of Index 1 (i7) and Index 2 (i5) indexes for each sample, where no index is ever used more than once. Illumina sequencers using patterned flowcell technology like HiSeq, NovaSeq, and iSeq can show higher incidences of misassigned index assignment or index hopping. Using UDI can prevent such misassignments when using those instruments for sequencing.
What is RNA strand specificity?
mRNA transcripts can come from either the plus or minus strand of gDNA template. Strand-specific RNA library preps, enable you to determine the original orientation of the transcripts while non-stranded library protocols lose this information and you cannot tell whether a sequencing read came from the plus or minus strand of the gDNA template.
How does the kit ensure strand specificity?
With non-stranded RNA library prep, RNA transcripts are fragmented and random primers are used to create the first- and second-strand synthesis of cDNA before any adapters are added or fragments are amplified. As a result, the first and second cDNA strands of the two antisense transcripts look exactly the same and cannot be distinguished when sequenced. The YourSeq workflow adds an adapter to the first strand of cDNA only, while the transcript is still a cDNA-RNA hybrid, using a DNA dependent DNA polymerase (DNA Pol I) which primes off the cDNA strand and cannot prime off the RNA strand.
What is 3'DGE?
For 3’-DGE RNA-seq libraries, the cDNA synthesis step uses an adapter with an oligo(dT) to prime off the transcript’s poly(A) tail. This means that 3’-DGE reads tend to map only to the 3’-end of the transcript. Since there is a theoretical 1-to-1 correlation between transcript molecule and cDNA molecule, transcript length is irrelevant and the number of reads mapping to a gene is solely a function of transcript abundance. A long gene, therefore, will have the same number of reads mapping to it as will a short gene of similar transcript abundance.
What are the advantages of 3'DGE RNA-seq?
The reduced complexity associated with 3’-DGE confers one of the biggest advantages. Transcript counting means that less sequencing depth is needed for a robust analysis compared to conventional RNA-seq. Also, paired-end sequencing does not add value for 3’-DGE. So, a researcher who might sequence conventional RNA-seq libraries at 20-30 million pairs of paired-end reads per sample can reduce cost by sequencing those same samples as 3’-DGE libraries at 3-10 million single reads per sample.
What are the limitations of 3'DGE RNA-seq?
Because 3'DGE RNA-seq only covers the 3'end of the transcript, it's not appropriate for detection of exonic polymorphisms, splice variant analysis, or the detection of gene fusion abundance. That kind of information can only be determined by sequencing the whole transcript.
What are the advantages of Full Transcript RNA-seq?
Full Transcript RNA-seq is required whenever a determination can only be made from the whole RNA transcript. It should be used for the detection of exonic polymorphisms, splice variant analysis, or the detection of gene fusion abundance.
What are the limitations of Full Transcript RNA-seq?
The only drawback with Full Transcript RNA-seq is the increased cost incurred because at least 20-30 million paired end reads are required per sample, due to the increased complexity of the library.
What type of RNA input can be used?
The YourSeq (FT & 3'DGE) Strand-Specific mRNA Library Prep Kits can be used with 5-100 ng of either poly A enriched or ribosomal depleted mRNA. Total RNA contains approximately 3-5% mRNA. For 5-100 ng mRNA we recommend starting with 100 ng - 5000 ng of total RNA.
What is rRNA depletion versus mRNA selection?
Ribosomal RNA (rRNA) depletion is a method that removes rRNA from total RNA, either during or after the isolation of total RNA so that the final result is enriched for mRNA. It can also be performed during library prep. Poly A enrichment uses oligo dT primers to selectively capture the polyadenylated 3' end of mRNA transcripts so that that the final result is enriched for mRNA.
How many sequencing reads are recommended?
For conventional full transcript (FT) RNA-seq libraries we recommend 20-30 million pairs of paired-end reads per sample. For 3’-DGE libraries we recommend 3-10 million single reads per sample.
Are there kit controls?
Each kit comes with drosophila melanogaster mRNA as a positive control.
YourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep Documents
YourSeq Indexes Library Prep (After March 2024)
- YourSeq Indexes Library Prep 24 CDI
- YourSeq Indexes Library Prep 24 UDI
- YourSeq Indexes Library Prep 96 CDI
- YourSeq Indexes Library Prep 96 UDI
YourSeq Indexes Library Prep (Through March 2024)
- YourSeq Indexes Library Prep 24 CDI
- YourSeq Indexes Library Prep 24 UDI
- YourSeq Indexes Library Prep 96 CDI
- YourSeq Indexes Library Prep 96 UDI
You might also be interested in:
| Name | Format | Cat No. | Price | |
|---|---|---|---|---|
| YourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep Kit 24 CDI | 24 rxns | 23001 | ¥8,390 | Add to Cart |
| YourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep Kit 96 CDI | 96 rxns | 23002 | ¥29,900 | Add to Cart |
| YourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep Kit 24 UDI | 24 rxns | 23003 | ¥10,140 | Add to Cart |
| YourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep Kit 96 UDI | 96 rxns | 23004 | ¥35,100 | Add to Cart |