理解并使用ATAC-Seq的完整指南

2021年2月9日
染色质免疫沉淀(ChIP)和RRBS等方法使研究人员更容易在全基因组范围内研究表观遗传修饰。 但是,这些方法的潜在局限性之一是您需要对什么表观遗传学机制在其中起作用有初步的想法。
ATAC-Seq向研究人员提供有关整个基因组中可及染色质的信息,而不涉及具体表观遗传学机制。 许多研究人员已开始使用ATAC-Seq作为初步筛查方法来鉴定样品之间可及染色质区域的变化,然后他们可以据此进行后续实验。
本文讨论了ATAC-Seq是什么,它的历史,工作原理以及使用ATAC-Seq发现的一些成果。
什么是ATAC-Seq?
ATAC-Seq是“Assay for Transposase-Accessible Chromatin with high-throughput Sequencing”的缩写。 ATAC-Seq方法依赖于使用高活性转座酶Tn5的下一代测序(NGS)文库的构建。将NGS接头连接到转座酶上,该转座酶可以使染色质断裂并同时将这些接头整合到开放的染色质区域中。构建的文库可通过NGS测序,并使用生物信息学分析具有可及或可访问染色质的基因组区域。
与其他技术(例如研究相似染色质特征的FAIRE-Seq或DNase-Seq)相比,ATAC-Seq的主要优势在于该测定所需的细胞数量更少,并且其两步法操作相对简单。

ATAC-Seq的历史
ATAC-Seq方法由斯坦福大学的Howard Chang和William Greenleaf实验室的首席研究员Jason Buenrostro于2013年在《Nature Methods》杂志上首次发表。
当时,他们正在寻找用于研究开放染色质,核小体定位和转录因子占用的新方法。现有的所有方法都有一些局限性,包括对大量起始原料的需求,复杂且耗时的方案,以及缺乏一次测定中同时评估上述三种染色质机理的能力。
当细胞数量过少使用现有技术无法分析时,研究人员团队着手克服当前方法的局限性,开发了新的技术分析表观遗传的整体面貌。
他们的方法依赖于已经用于基于标签的NGS文库制备方法的高活性Tn5转座酶,例如Epicenter开发的Nextera方法(后来被Illumina收购)。标记过程可用于体外裂解基因组DNA,并同时添加用于高通量测序的接头。作者假设,如果在体内使用类似的方法,则接头的添加将主要发生在开放的染色质区域中,在该区域中不会发生转座酶的空间位阻,从而允许该酶优先进入这些区域。
在概念验证研究中,他们研究了从500和50,000个细胞群体中分离出的未固定细胞核。他们生成的数据与DNase-Seq和FAIRE-Seq非常相似,而两者都需要1到5千万个细胞。这表明ATAC-Seq方法在所需样品数量方面比那些较旧的方法有了显著改进。
此外,由于转座酶仅将NGS接头添加到开放的DNA中,因此ATAC-Seq具有另一个好处:它还可用于生成核小体位置以及转录因子结合的高分辨率图谱。相对较短的两步操作方案表明ATAC-Seq也有潜力用于从临床样品中生成个人表观遗传概况。

ATAC-Seq如何工作?
如前所述,与其他分析开放染色质区域或核小体定位的方法(例如DNase-Seq或FAIRE-Seq)相比,ATAC-Seq的主要优势之一是可以用非常少的细胞(约50,000个细胞)在几个小时内分两步完成实验。相比之下,传统方法更加费力且需要数百万个细胞。
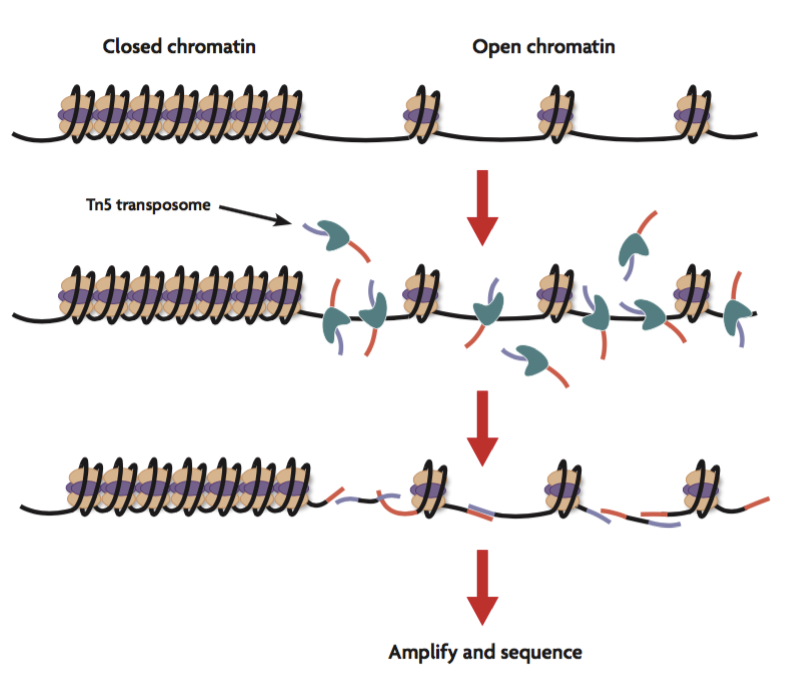
下面,我们简要概述了ATAC-Seq实验步骤,该步骤可分为两个主要部分:细胞制备和转座。
细胞制备
第一步需要收获细胞。由于用于ATAC-Seq分析的细胞数量对于转座反应和生成的DNA片段的大小分布至关重要,因此对细胞进行计数非常重要。此外,细胞应完好无损,并且是均匀的单细胞悬浮液。收获后,用非离子型去污剂裂解细胞以产生纯细胞核。
转座反应
然后将所得的染色质片段化,并同时使用Tn5转座酶进行测序接头标记,以生成ATAC-Seq文库。纯化后,可使用barcode引物通过PCR扩增文库。所得文库随后通过qPCR或NGS进行分析。
其他ATAC-Seq注意事项
使用正确的细胞数量用于转座是很重要的。一般建议使用25,000-75,000个细胞。使用太少或太多的细胞会导致消化过度或消化不足,这会降低文库的质量。此外,细胞的固定和剧烈的机械剪切趋于降低数据质量。
最近的ATAC-Seq应用和突破
通过短短的两步过程,ATAC-Seq已被证明适用于各种细胞类型和模式系统。自该方法于2013年发布以来,使用ATAC-Seq发表的论文数量每年增加一倍。我们在下面描述了来自不同领域的一些文献,显示了该方法的影响和适用性。
ATAC-Seq与癌症
迈阿密的一组研究人员及其合作者在不断发展的癌症表观遗传学领域中使用了ATAC-Seq,以研究Polycomb Repressed Complex 1(PRC1)对乳腺癌中染色质状态的影响。 ATAC-Seq实验表明,RING1B(PRC1复合物的一个成员)的降低会导致增强子区域的染色质可及性发生变化。此外,RING1B的丢失导致约500个开放的染色质峰的丢失,并且在增强子附近出现了600多个新的开放区域。Motif分析表明,增强子区域丢失的峰包含FOXA1 / 2结合位点,而预测的峰包含CTCF结合位点,表明RING1B在乳腺癌细胞中具有双重作用。

使用ATAC-Seq研究衰老
年龄相关性黄斑变性(AMD)是一种导致不可逆视力下降的疾病。虽然已知有几个遗传基因位点会增加这种疾病的风险,但表观遗传因素的作用在很大程度上尚不清楚。
约翰霍普金斯大学医学院的科学家使用ATAC-Seq来研究AMD与对照患者的全基因组染色质开放性。他们的结果表明,AMD患者的视网膜和视网膜色素上皮细胞(RPE)的染色质开放性大大降低。进一步分析数据表明,特定的转录因子和细胞功能在差异可及染色质区域富集。
而且,他们研究了潜在的环境影响,例如香烟烟雾对AMD的影响。用香烟烟雾处理末端分化的iPSC衍生的RPE细胞可导致染色质可及性下降,类似于AMD患者中观察到的模式。
最后,研究人员试图确定治疗AMD的潜在药物靶标。对RNA-Seq数据的分析表明,组蛋白脱乙酰基酶HDAC11在AMD中过表达,可能成为未来治疗的靶标。
ATAC-Seq和免疫学
在体液免疫应答中的成熟期间,B细胞祖细胞需要经历大量的表型变化,这种变化是由涉及多个转录因子协调作用的多层染色质组织介导的。康奈尔大学的一组研究人员及其合作者发表在《Nature Immunology》上的一项最新研究调查了赖氨酸特异性脱甲基酶1(LSD1)在B细胞发育中的作用。
该团队使用ATAC-Seq研究了Lsd1突变小鼠中分选的存活生发中心B细胞中的开放染色质区域。他们观察到733个区域在Lsd1敲除后获得了染色质的可及性,而314个区域失去了可及性。与这733个开放区域相关的转录激活也增加了。进一步的实验,包括ChIP-Seq,揭示了一种对增强子和启动子停顿的协调很重要的转录抑制物,BCL6,直接结合LSD1蛋白并将其募集到染色质上,而这种相互作用是驱动生发中心B细胞恶性转化所必需的。
使用ATAC-Seq研究细胞分化和发育
细胞分化和谱系定型通常由表观遗传机制介导,包括染色质结构的大变化以及从祖细胞和干细胞中通常更开放的染色质状态逐渐过渡到分化细胞中更封闭的紧密染色质状态。
在《Science》杂志上发表的一篇论文中,以色列的一组研究人员研究了血细胞发育过程中的染色质状态。他们通过观察在所有主要血细胞谱系发育的不同阶段的四个组蛋白修饰(H3K4me1,H3K4me2,H3K4me3和H3K27ac)来表征造血作用的分化阶段。
使用iChIP,他们能够识别48,415种增强子,其中的90%在造血过程中状态发生了变化。这些增强子中有60%表现出典型的行为,即它们逐渐失活并仅在相关谱系中保持活性,而约40%在分化过程中被新激活。
这些新建立的增强子包含H3K4me1修饰,这与染色质可及性的增加有关。为了测量这些增强子上的染色质是否更可及,将H3K4me1和H3K27ac ChIP-Seq数据与ATAC-Seq数据相关联。研究人员观察到,与H3K27ac相比,ATAC-Seq峰与H3K4me1的重叠具有更紧密的相关性,这表明增强子处H3K4me1的得失会影响可及染色质的形成。此外,ATAC-Seq能够识别造血过程中新的血细胞谱系调控因子。
总而言之,ATAC-Seq使科学家能够清楚地证明染色质状态在血细胞分化过程中非常动态,增强子不仅如先前报道的那样失活,而且从头建立,这导致了大规模动态重组的新模型分化和发育过程中染色质的变化。
单细胞ATAC-Seq(scATAC-Seq)
使用数百万个细胞群体的所有方法的主要局限性在于,它们产生的数据始终是该群体中每个细胞的平均值,这抹平了可能存在的细胞间差异,因此可能会消除对子种群中有趣现象进行观察的能力。当涉及诸如肿瘤异质性或发育过程之类时,这种可变性通常是生物学的重要特征。ATAC-Seq的原始发明者,Jason Buenrostro和他的同事,在《Nature》发文表示正着手通过修改ATAC-Seq的实验步骤使其适应单细胞分析。
他们最初的实验使用了Fluidigm的微流体平台来帮助他们研究单细胞。但是当今许多单细胞ATAC-Seq(scATAC-Seq)实验步骤都使用10X Genomics平台。 分离单个细胞后,将它们用Tn5转座酶标记,并使用具有细胞识别条形码的引物通过PCR扩增文库。 合并这些文库后,即可照常对其进行测序。
总而言之,这种方法改进了ATAC-Seq,使研究人员能够在单细胞水平上研究许多样本类型的生物学变异,这将使人们对细胞异质性的分子基础有更深入的了解。

如何在研究中开始使用ATAC-Seq
要在您的实验室开始使用ATAC-Seq,您可以向Active Motif咨询。我们提供了一站式的ATAC-Seq服务。您将样本(细胞或组织)发送给我们,我们会在数周内提供您分析数据。 ATAC-Seq服务涵盖实验中的所有步骤,包括膜通透,添加带有接头的转座酶,在开放基因组位置通过转座酶插入接头,文库扩增,Illumina平台上的NGS以及生物信息学分析。我们对标准ATAC-Seq实验进行了优化,以确保我们将为您生成最优质的数据。
我们接受培养的细胞系,原代细胞或冷冻组织样品,建议提交100,000个细胞或20-50 mg冷冻组织。重复次数始终是一个有争议的问题。对于我们的ATAC-Seq服务,因为我们已经证明生成的数据的可重复性非常好,通常只建议两次重复。但是,最终您将决定最佳实验设计。
生物信息学数据分析由我们内部的生物信息学专家团队进行。标准的生物信息学分析包括:
- 序列分析:将测序读段比对到基因组,并删除重复的reads。
- 峰发现: 使用MACS 2.1.0,将双端测序中的两个reads用于peak calling。
- 片段密度的确定: 为了鉴定基因组的转座事件的密度,将基因组分为32 bp的条带,并确定每个条带中的片段数。为此,将reads扩展到200 bp以使数据平滑。
- 通过随机采样对所有样本的比对reads数进行归一化,以使每一个样本包含所有样本中比对reads数最少的样本相同数目的reads。
- 活动区域分析: 为了比较两个或多个样本之间的峰,将重叠的峰分组为“活动区域”,这是由最上游峰的起始坐标和最下游峰的终止坐标定义的专有度量。
如果您正在处理复杂或异质的样品,则可以使用我们的单细胞ATAC-Seq服务,它可能是帮助您实现实验目标的最佳选择。
在您的实验室中建立ATAC-Seq分析也是一种选择。要自己进行ATAC-Seq,您首先需要获取预处理的Tn5转座酶。之后,您需要从细胞或组织中纯化细胞核,并为转座反应找到合适的条件(例如,染色质与Tn5之间的比例正确)。文库制备和扩增后,需要进行NGS,随后需要分析数据。尽管ATAC-Seq比以前的方法更快,更容易,但是它仍然给许多没有做过该实验的研究人员带来了挑战,导致科学家越来越多地向Active Motif的表观遗传学专家求助,以生成他们可以信赖的高质量数据。
总结:ATAC-Seq方法改变了研究人员进行表观遗传学研究的方式
当表观遗传学领域第一次起步时,各种各样的方法可能会令人不知所措。选择一种方法来解决您的生物学问题可能并不那么明显或容易。诸如DNase-Seq,FAIRE-Seq,DamID,Hi-C或ChIP-Seq之类的方法需要数百万个细胞,并且通常需要进行大量的优化。最后,它们可能只能传递有限的信息,使您从样本中获得的表观遗传学信息不完整。
ATAC-Seq改变了科学家入门表观遗传学的方式。 ATAC-Seq实验仅需要约50,000个细胞作为起始样本,并且其相对较短的两步实验,是开始表观遗传过程的一种有吸引力的方法。
无论您是要分析样品中染色质的状态还是比较特殊处理前后的染色质状态,ATAC-Seq都可以让您研究全基因组染色质的变化,并可以提供相关指导如应该进行哪些表观遗传修饰或转录因子的研究,以及在后续实验中采用哪种方法。
ATAC-Seq使得研究染色质及进入表观遗传学的第一步比以往任何时候都更加轻松快捷。
如果您有兴趣了解有关ATAC-Seq,我们的服务或获得报价的更多信息,请与我们联系。
About the author

Stefan Dillinger, Ph.D.
Stefan was born in the Free State of Bavaria, Germany. After studying biochemistry in Ulm and Regensburg, he got his Ph.D. in the field of epigenetics, studying the distribution of heterochromatin around nucleoli during cellular senescence. As a graduate student he started his own German science podcast “The Random Scientist” and is now the host of Active Motif’s Epigenetics Podcast. When Stefan is not working at Active Motif or recording podcasts, he is a passionate runner (he finished the New York City Marathon in 3 hours 21 minutes!!) and loves to spend time with his wife and son.
Contact Stefan on LinkedIn with any questions, or to get running advice.
Related Articles
Guide to Generating the Best ChIP Data

March 15, 2019
The chromatin immunoprecipitation (ChIP) assay has become one of the most popular laboratory techniques to investigate the association of DNA-binding proteins and histones with chromatin. This article covers the major challenges of ChIP assays and how to overcome them to generate the best ChIP data.
Read More
Comprehensive Guide to Understanding and Using CUT&Tag Assays

March 9, 2020
CUT&Tag shows a lot of promise and has the potential to alleviate some ChIP limitations, but it also has its own set of limitations that must be considered. This article covers what CUT&Tag is and describes the advantages and drawbacks of this method.
Read More
<< Back to MOTIFvations Blog Home Page




